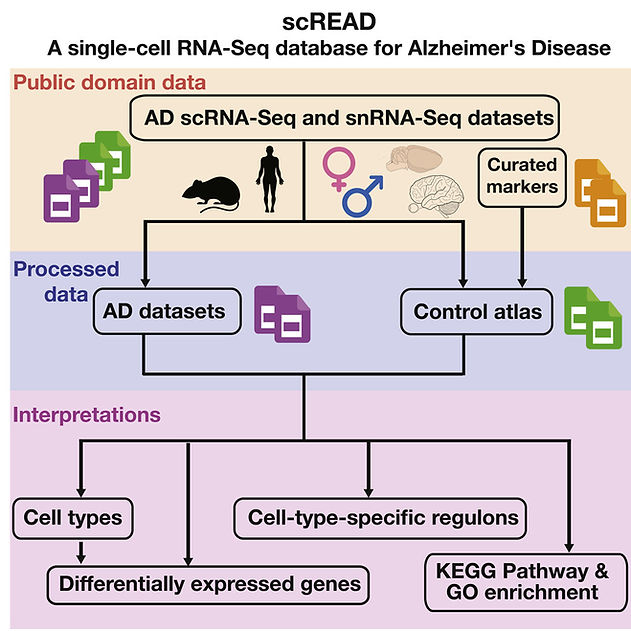
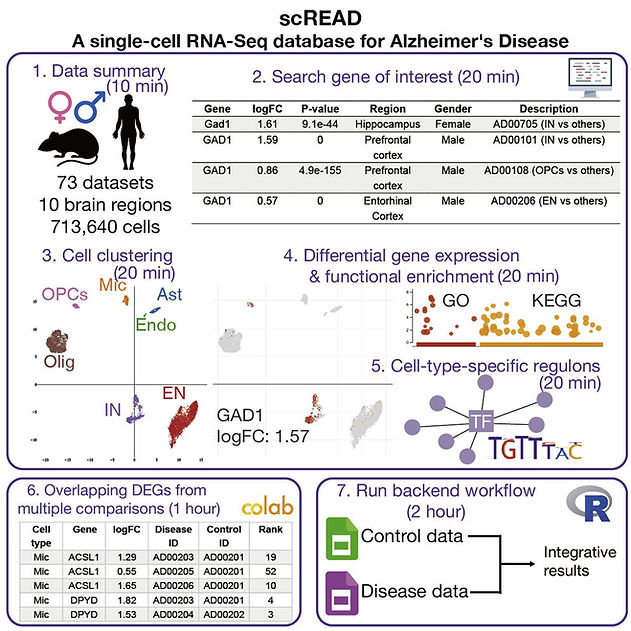
Alzheimer's disease (AD) is a progressive neurodegenerative disorder of the brain and the most common form of dementia among the elderly. The single-cell RNA-sequencing (scRNA-Seq) and single-nucleus RNA-sequencing (snRNA-Seq) techniques are extremely useful for dissecting the function/dysfunction of highly heterogeneous cells in the brain at the single-cell level, and the corresponding data analyses can significantly improve our understanding of why particular cells are vulnerable in AD. We developed an integrated database named scREAD (single-cell RNA-Seq database for Alzheimer's disease), which is as far as we know the first database dedicated to the management of all the existing scRNA-Seq and snRNA-Seq data sets from the human postmortem brain tissue with AD and mouse models with AD pathology. scREAD provides comprehensive analysis results for 73 data sets from 10 brain regions, including control atlas construction, cell-type prediction, identification of differentially expressed genes, and identification of cell-type-specific regulons.
Single-cell RNA-sequencing (scRNA-seq) and single-nucleus RNA-sequencing (snRNA-seq) studies have provided remarkable insights into understanding the molecular pathogenesis of Alzheimer's disease. We recently developed scREAD, a database to provide comprehensive analyses of all the existing AD scRNA-seq and snRNA-seq data from the public domain. Here, we report protocols for using the scREAD web interface and running the backend workflow locally. Our protocols enable custom analyses of AD single-cell and single-nucleus gene expression profiles.

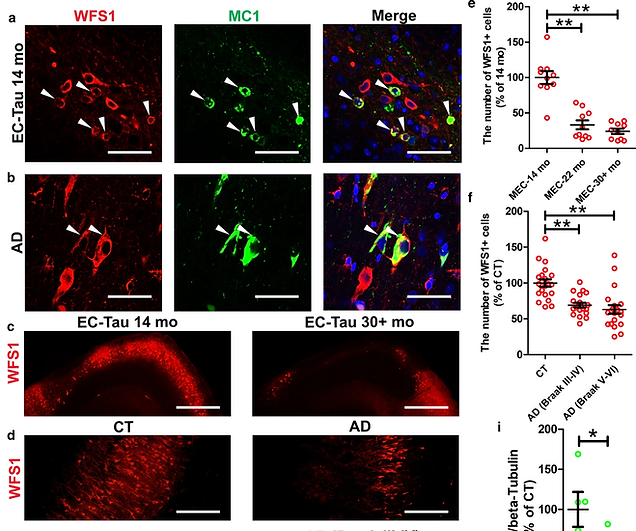
Selective neuronal vulnerability to protein aggregation is found in many neurodegenerative diseases including Alzheimer’s disease (AD). Understanding the molecular origins of this selective vulnerability is, therefore, of fundamental importance. Tau protein aggregates have been found in Wolframin (WFS1)-expressing excitatory neurons in the entorhinal cortex, one of the earliest affected regions in AD. The role of WFS1 in Tauopathies and its levels in tau pathology-associated neurodegeneration, however, is largely unknown. Here we report that WFS1 deficiency is associated with increased tau pathology and neurodegeneration, whereas overexpression of WFS1 reduces those changes. We also find that WFS1 interacts with tau protein and controls the susceptibility to tau pathology. Furthermore, chronic ER stress and autophagy-lysosome pathway (ALP)-associated genes are enriched in WFS1-high excitatory neurons in human AD at early Braak stages. The protein levels of ER stress and autophagy-lysosome pathway (ALP)-associated proteins are changed in tau transgenic mice with WFS1 deficiency, while overexpression of WFS1 reverses those changes. This work demonstrates a possible role for WFS1 in the regulation of tau pathology and neurodegeneration via chronic ER stress and the downstream ALP. Our findings provide insights into mechanisms that underpin selective neuronal vulnerability, and for developing new therapeutics to protect vulnerable neurons in AD.
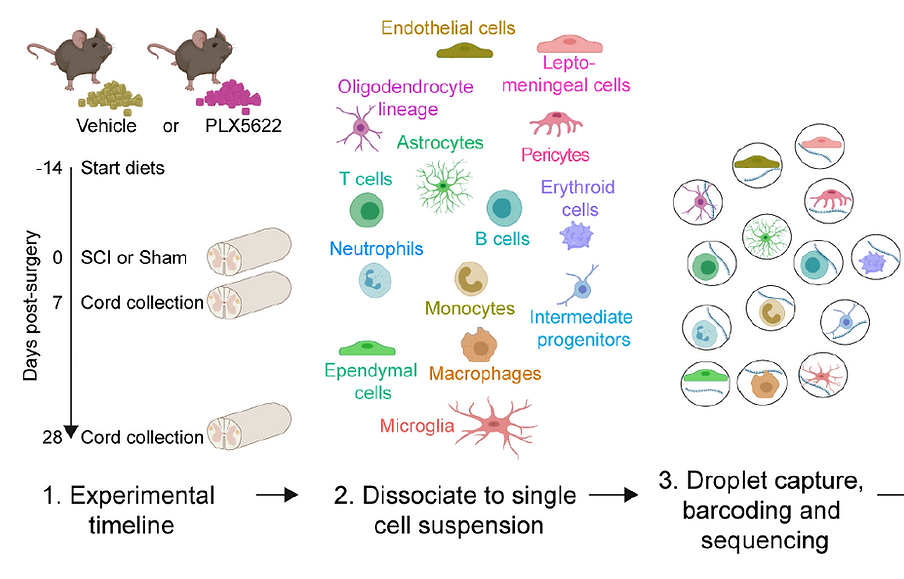
Traumatic spinal cord injury (SCI) triggers a neuro-inflammatory response dominated by tissue-resident microglia and monocyte derived macrophages (MDMs). Since activated microglia and MDMs are morphologically identical and express similar phenotypic markers in vivo, efforts to identify the predominant roles that microglia play in SCI have historically been challenging. However, with the advent of new research tools, key roles for microglia in regulating diverse post-injury cellular responses are emerging. Here, we pharmacologically depleted microglia and used a comprehensive array of anatomical, histopathological, tract tracing, bulk and single cell RNA sequencing techniques to reveal the cellular and molecular responses to SCI that are controlled by microglia. New data indicate that microglia are vital for SCI recovery and coordinate injury responses in CNS-resident glia and infiltrating leukocytes. Indeed, depleting microglia exacerbated tissue damage and worsened recovery of function. Conversely, restoring select microglia-dependent signaling axes, identified through bioinformatic analysis of sequencing data, in microglia depleted mice prevents secondary damage and promotes functional recovery. Additional bioinformatics analyses reveal that optimal repair after SCI and likely other forms of neurological disease, might be achieved by co-opting key ligand-receptor interactions between microglia, astrocytes and MDMs.


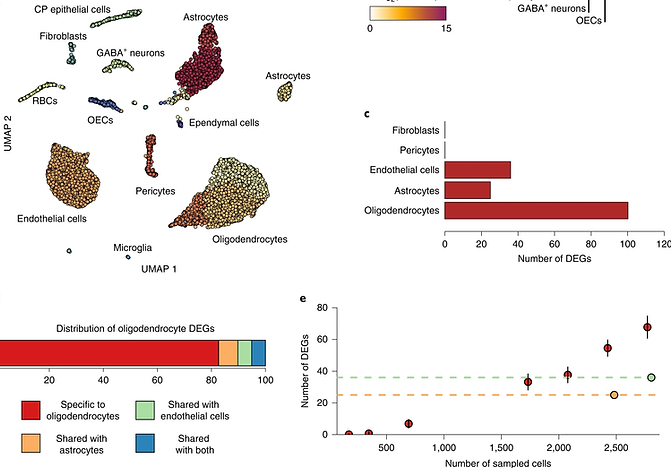
Alzheimer’s disease (AD) is a complex neurodegenerative disease, perturbing neuronal and non-neuronal cell populations. In this study, using single-cell transcriptomics, we mapped all non-immune, non-neuronal cell populations in wild-type and AD model (5xFAD) mouse brains. We identified an oligodendrocyte state that increased in association with brain pathology, which we termed disease-associated oligodendrocytes (DOLs). In a murine model of amyloidosis, DOLs appear long after plaque accumulation, and amyloid-beta (Aβ) alone was not sufficient to induce the DOL signature in vitro. DOLs could be identified in a mouse model of tauopathy and in other murine neurodegenerative and autoimmune inflammatory conditions, suggesting a common response to severe pathological conditions. Using quantitative spatial analysis of mouse and postmortem human brain tissues, we found that oligodendrocytes expressing a key DOL marker (SERPINA3N/SERPINA3 accordingly) are present in the cortex in areas of brain damage and are enriched near Aβ plaques. In postmortem human brain tissue, the expression level of this marker correlated with cognitive decline. Altogether, this study uncovers a shared signature of oligodendrocytes in central nervous system pathologies.
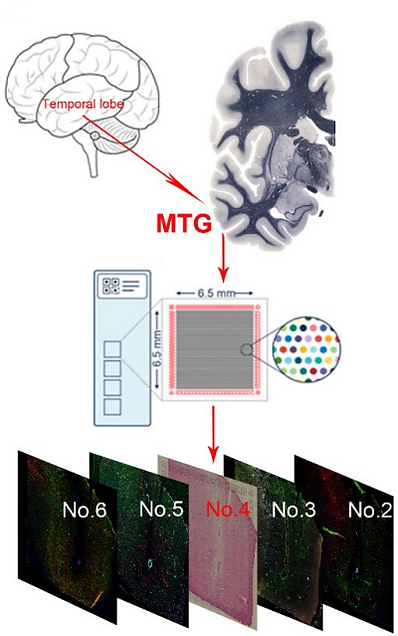
Alzheimer’s disease (AD) is pathologically characterized by amyloid beta (Aβ) plaques, neurofibrillary tangles (tau aggregates), and alterations in microglia, astrocytes and oligodendrocytes. The mesial temporal lobe is a vulnerable brain region in early AD; however, little is known about the transcriptome-scale gene expression in this region and its relation to AD pathology. Here we use the 10x Genomics Visium platform in combination with co-immunofluorescence staining of AD-associated pathological markers to define the spatial topography of gene expression in the middle temporal gyrus (MTG) from both early AD and age- and gender-matched control cases. We identify unique marker genes for six cortical layers and the adjacent white matter as well as gene expression patterns and alterations that showcase unique gene signatures and pathways associated with a range of AD pathology. Also, gene co-expression analyses of differentially expressed genes (DEGs) between AD and controls reveal four unique gene modules, which significantly change their co-expression patterns in the presence of variations of AD pathology. Furthermore, we validate the changes of key representative DEGs that are associated with AD pathology in neurons, microglia, astrocytes and oligodendrocytes using single-molecule fluorescent in situ hybridization. In summary, we provide a rich resource for the spatial transcriptomic profile of the human MTG, which will contribute to our understanding of the complex architecture and AD pathology of this vulnerable brain regions.