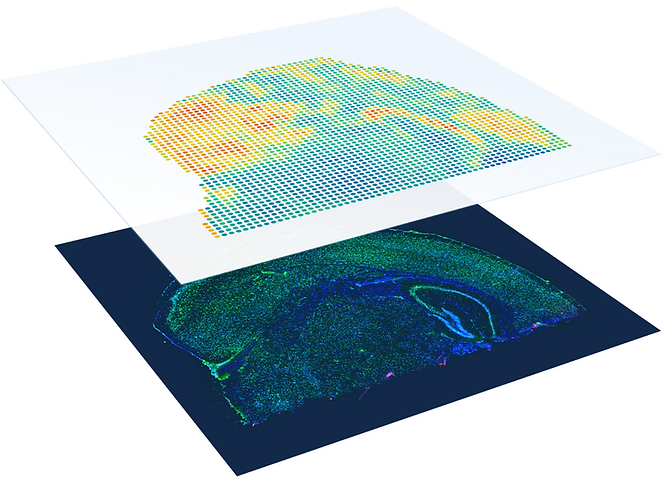

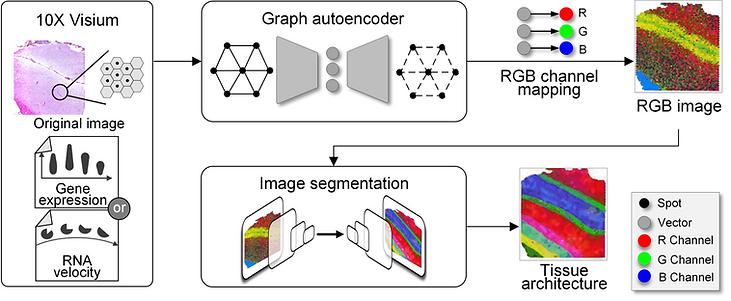
Spatially resolved transcriptomics provides a new way to define spatial contexts and understand the pathogenesis of complex human diseases. Although some computational frameworks can characterize spatial context via various clustering methods, the detailed spatial architectures and functional zonation often cannot be revealed and localized due to the limited capacities of associating spatial information. We present RESEPT, a deep-learning framework for characterizing and visualizing tissue architecture from spatially resolved transcriptomics. Given inputs such as gene expression or RNA velocity, RESEPT learns a three-dimensional embedding with a spatial retained graph neural network from spatial transcriptomics. The embedding is then visualized by mapping into color channels in an RGB image and segmented with a supervised convolutional neural network model. Based on a benchmark of 10x Genomics Visium spatial transcriptomics datasets on the human and mouse cortex, RESEPT infers and visualizes the tissue architecture accurately. It is noteworthy that, for the in-house AD samples, RESEPT can localize cortex layers and cell types based on pre-defined region- or cell-type-enriched genes and furthermore provide critical insights into the identification of amyloid-beta plaques in Alzheimer's disease. Interestingly, in a glioblastoma sample analysis, RESEPT distinguishes tumor-enriched, non-tumor, and regions of neuropil with infiltrating tumor cells in support of clinical and prognostic cancer applications.

High throughput spatial transcriptomics (HST) is a rapidly emerging class of experimental technologies that allow for profiling gene expression in tissue samples at or near single-cell resolution while retaining the spatial location of each sequencing unit within the tissue sample. Through analyzing HST data, we seek to identify sub-populations of cells within a tissue sample that may inform biological phenomena. Existing computational methods either ignore the spatial heterogeneity in gene expression proles, fail to account for important statistical features such as skewness, or are heuristic-based network clustering methods that lack the inferential benefits of statistical modeling. To address this gap, we develop SPRUCE: a Bayesian spatial multivariate finite mixture model based on multivariate skew-normal distributions, which is capable of identifying distinct cellular sub-populations in HST data. We further implement a novel combination of Polya-Gamma data augmentation and spatial random effects to infer spatially correlated mixture component membership probabilities without relying on approximate inference techniques. An R package spruce for fiting the proposed models is available through The Comprehensive R Archive Network (CRAN).

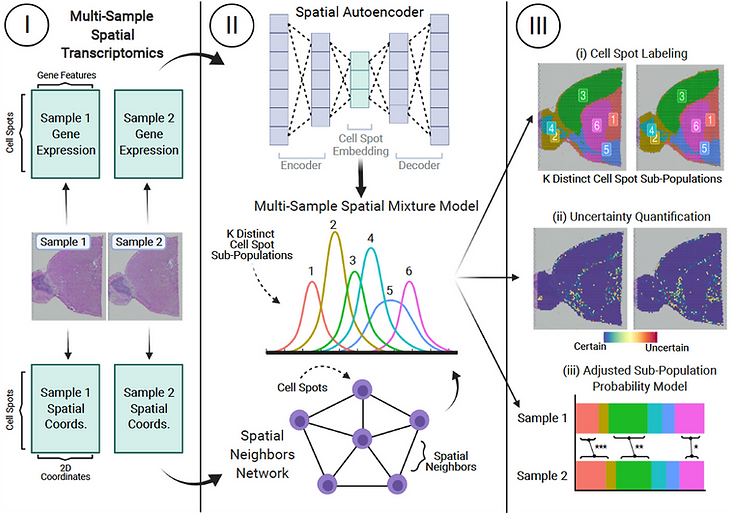
High throughput spatial transcriptomics (HST) technologies have allowed for identification of distinct cell sub-populations in tissue samples, i.e., tissue architecture identification. However, existing methods do not allow for simultaneous analysis of multiple HST samples. Moreover, standard tissue architecture identification approaches do not provide uncertainty measures. Finally, no existing frameworks have integrated deep learning with Bayesian statistical models for HST data analyses. To address these gaps, we developed MAPLE: a hybrid deep learning and Bayesian modeling framework for detection of spatially informed cell spot sub-populations, uncertainty quantification, and inference of group effects in multi-sample HST experiments. MAPLE includes an embedded regression model to explain cell sub-population abundance in terms of available covariates such as treatment group, disease status, or tissue region. We demonstrate the capability of MAPLE to achieve accurate, comprehensive, and interpretable tissue architecture inference through four case studies that spanned a variety of organs in both humans and animal models.
.jpg)
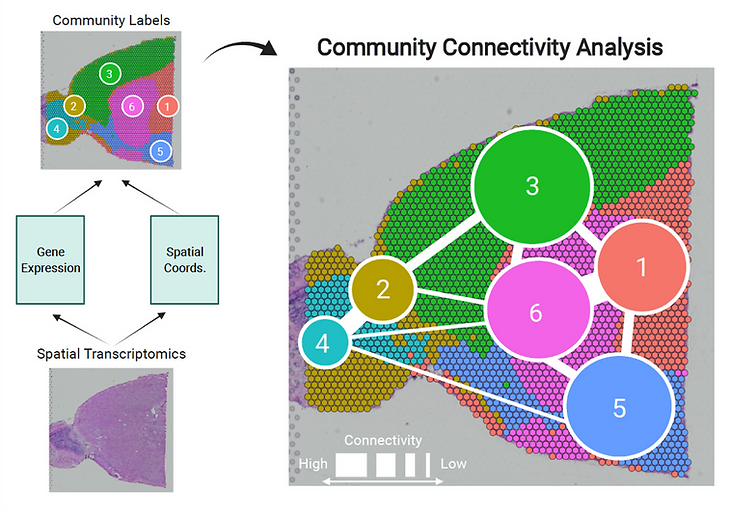
The advent of high throughput spatial transcriptomics (HST) has allowed for unprecedented characterization of spatially distinct cell communities within a tissue sample. While a wide range of computational tools exist for detecting cell communities in HST data, none allow for characterization of community structure – an analysis task that can help elucidate cellular dynamics in important settings such as the tumor microenvironment. To address this gap, we introduce the concept of community connectivity analysis (CCA), which is concerned not only with labeling distinct cell communities within a tissue sample, but understanding the relative similarity of cells within and between communities. We develop a Bayesian multi-layer network model called BANYAN for integration of spatial and gene expression information to achieve CCA. We use BANYAN to implement CCA in invasive ductal carcinoma, and uncover distinct community structure relevant to the interaction of cell types within the tumor microenvironment. Next, we show how CCA can help clarify ambiguous annotations in a human white adipose tissue sample. Finally, we demonstrate improved community detection with BANYAN as a result of considering spatial information via a simulation study based on real sagittal mouse brain HST data.
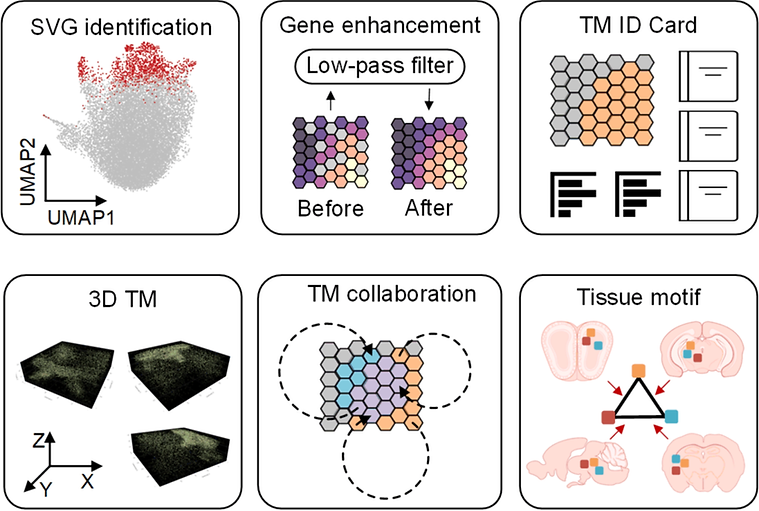
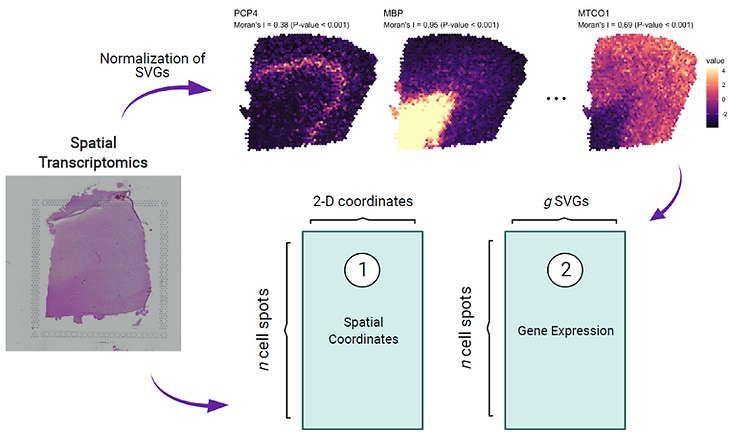
The tissue module (TM) was defined as an architectural area containing recurrent cellular communities executing specific biological functions at different tissue sites. However, the computational identification of TMs poses challenges owing to their various length scales, convoluted biological processes, not well-defined molecular features, and irregular spatial patterns. Here, we present a hypothesis-free graph Fourier transform model, SpaGFT, to characterize TMs. For the first time, SpaGFT transforms complex gene expression patterns into simple, but informative signals, leading to the accurate identification of spatially variable genes (SVGs) at a fast computational speed. Based on clustering the transformed signals of the SVGs, SpaGFT provides a novel computational framework for TM characterization. Three case studies were used to illustrate TM identities, the biological processes of convoluted TMs in the lymph node, and conserved TMs across multiple samples constituting the complex organ. The superior accuracy, scalability, and interpretability of SpaGFT indicate that it is a novel and powerful tool for the investigation of TMs to gain new insights into a variety of biological questions.