FAQ
This list of questions is not exhaustive. If you have any other questions please email any authors on the Contact page.
How to cite ssREAD?
ssREAD is still under review.
What is Alzheimer’s disease?
Alzheimer’s disease (AD) is the most common cause of dementia, accounting for an estimated 60% to 80% of cases. Currently, there are an estimated 5.8 million Americans age 65 or older suffering from AD. Difficulty in remembering recent conversations, names, or events is often an early clinical symptom. The hallmark pathologies of Alzheimer’s disease are the accumulation of the protein fragment beta-amyloid (plaques) outside neurons in the brain and twisted strands of the protein tau (tangles) inside neurons. These changes are accompanied by the death of neurons and damage to brain tissue. Alzheimer’s is a slowly progressive brain disease that begins many years before symptoms emerge. Unfortunately, there are no effective therapeutics for the prevention and treatment of the disease. (2020 Alzheimer’s Disease Facts and Figures)
What is single-cell RNA sequencing?
Single-cell RNA sequencing (scRNA-Seq) examines the dynamic transcriptomic profile of individual cells with next-generation sequencing technologies, and hence provide a higher resolution of cellular differences and a better understanding of the function of an individual cell in the context of its microenvironment. The scRNA-Seq can reveal complex and rare cell populations, uncover regulatory relationships between genes, and track the trajectories of distinct cell lineages in development. The scRNA-Seq is becoming widely used across biological disciplines including neuroscience. More recently, scRNA-Seq has been used in the field of AD and significantly improves our understanding of the pathogenesis of AD at the single-cell level.
What is spatial transcriptomics?
Spatial transcriptomics is a groundbreaking molecular profiling method that allows scientists to measure all the gene activity in a tissue sample and map where the activity is occurring. Already this technology is leading to new discoveries that are proving instrumental in helping scientists gain a better understanding of biological processes and disease.
What is ssREAD?
ssREAD (single-cell and spatial RNA-Seq database for Alzheimer’s Disease), dedicated to the management of scRNA-Seq, single-nucleus RNA-sequencing (snRNA-Seq), and Spatial transcriptomics datasets from human postmortem brain tissue with AD and mouse models with AD pathology. ssREAD has major updates to include more datasets and enhanced analytical pipeline, visibility, and usability:
-
ssREAD hosts comprehensive scRNA-seq & snRNA-seq & ST datasets of AD and their analysis results, which are not covered by any single existing database or resource. Specifically, we include 172 datasets collected from 34 AD-related scRNA-seq & snRNA-seq studies, covering 2,194,144 cells. In addition, 300 ST datasets collected from 12 brain studies (two AD studies and ten healthy brain studies) are collected and archived in ssREAD. All datasets are well-annotated with species, gender, brain region, disease/control, age, and pathology stage.
-
ssREAD provides in-depth analysis, such as cell clustering and annotation, identification of cell-type-specific marker genes, differentially expressed genes (DEGs), spatially variable genes (SVGs), and cell-type-specific regulons, for both individual data and integrated cell atlas. All results interpretations are under the guidance of Dr. Hongjun (Harry) Fu, who is an Assistant Professor in the Ohio State University with expertise in AD studies, especially the selective cellular and regional vulnerability. We also provide cross-data queries and analyses, such as DEGs between males and females across multiple datasets. Specifically, we provide spot deconvolution for the integrative analysis of ST and scRNA-seq & snRNA-seq data.
-
All the analytical results are presented through a one-stop and user-friendly interface to free AD biologists from programming burdens. New interactive plots are provided for visualizing cells and spatial spots, including scatter plots, bar plots, and violin plots. Real-time analyses are available for the query of DEGs, SVGs, and functional enrichment analysis.
-
All datasets, including the metadata & cell type labels from the authors, are benchmark-ready and can be downloaded as different object formats (e.g., .h5ad and .h5seurat), so that they can be easily loaded and analyzed by Seurat and Scanpy.
What is a spatially variable gene (svg)?
SVGs are spatial dependence for a given gene by testing whether gene expression levels at different locations covary in a manner that depends on their relative location, and thus are spatially variable.
What is deconvolution analysis?
Deconvolution is a computational method that treats the image as an estimate of the true specimen intensity and using an expression for the point spread function performs the mathematical inverse of the imaging process to obtain an improved estimate of the image intensity. It track compositional alterations of cell types (scRNA-seq or snRNA-seq dataset) in gene expression data (spatial transcriptomics dataset).
Which browsers are supported?
The browsers that ssREAD supported are Google Chrome, Safari, and Firefox. The ssREAD is not supported by the Internet Explorer browser.
Which species are covered in ssREAD?
Human and Mouse.
How many data files are covered?
The ssREAD contains 34 existing human and mouse AD scRNA-Seq & snRNA-Seq datasets, and 12 spatial transcriptomic studies from the public domain. These files are redefined according to species, gender, brain region, disease or control, and age.
How can we define different mouse age stages?
The choice of rodent age should be given more consideration, as it improves the quality and relevance of the data obtained, and reduces variability. As for the age of mice used, it depends on the development or senescence of the system being studied. For example, Flurkey et al. defined a mature adult C57BL6/J mouse as 3–6 months (equivalent in this analysis to 20–30 human years), a middle-aged mouse as 10–14 months (equivalent to 38–47 human years) and an old mouse as 18–24 months (equivalent to 56–69 human years). We, therefore, define the different mouse age stages in our database as shown in the following table:
| Age_Stage | Range of ages |
|---|---|
| 2 months | 1-2 months |
| 7 months | 4-7 months |
| 15 months | 10-15 months |
| 20 months | 20 months |
Where can I find subclusters?
The subcluster information can be found at the ‘Cell Clustering’ part. For each cell type, we have done the subcluster finding analysis.
What are the evaluation indexes for identified cell types?
- If benchmark labels are provided from the original study, the identified cell labels will be evaluated by the Adjusted Rand Index (ARI).
- If benchmark labels are not provided from the original study, the predicted cell types will be evaluated by calculating the silhouette score that measures how similar a cell is to its type compared to other clusters.
What is differential gene expression analysis?
We categorized the experiments for differential gene expression as one of the following conditions: AD versus control and AD versus AD in the same species under the same gender, brain region, and age. The differential gene expression analysis under each condition can be accessed through the Seurat FindMarkers function. All results were scaled by cell types and presented in the tables, allowing users to explore the differential expression of interesting genes across different conditions.
The cases how to achieve differential expressed genes between different conditions (Condition 1 v.s. Condition 2) for diverse cell types in ssREAD are showed in the following table:
| Species | If in the same region | Condition 1 | Condition 2 |
|---|---|---|---|
| Human | Yes | Disease | Control |
| Mouse | Yes | Disease | Control |
| Human | Yes | Disease | Disease |
| Mouse | Yes | Disease | Disease |
| Human | No | Disease | Disease |
| Mouse | No | Disease | Disease |
Note: The comparisons are all in the same gender and age.
What is enrichment analysis?
Enrichment analysis is a computational method for inferring knowledge about an input gene set by comparing it to annotated gene sets representing prior biological knowledge. Enrichment analysis checks whether an input set of genes significantly overlaps with annotated gene sets. For more information, you can read this excellent review about enrichment analysis here.
What are regulons and cell-type-specific regulons (CTSRs) and why are they important?
The term regulon was first introduced by Maas et al. in 1964, intended to name a maximal group of co-regulated genes, which may be scattered in a genome without apparent patterns in terms of their genomic locations.Based on that, cell-type-specific regulons (CTSRs) are defined as a group of genes, which receive similar regulatory signals in a specific cell type, hence tending to have similar expression patterns and share conserved motifs in this cell type. A successful elucidation of CTSRs will substantially improve the identification of transcriptionally co-regulated gene modules, realistically allowing reliable prediction of global transcription regulation networks encoded in a specific cell type.
How to interpret the CTSRs?
- Regulon heatmap
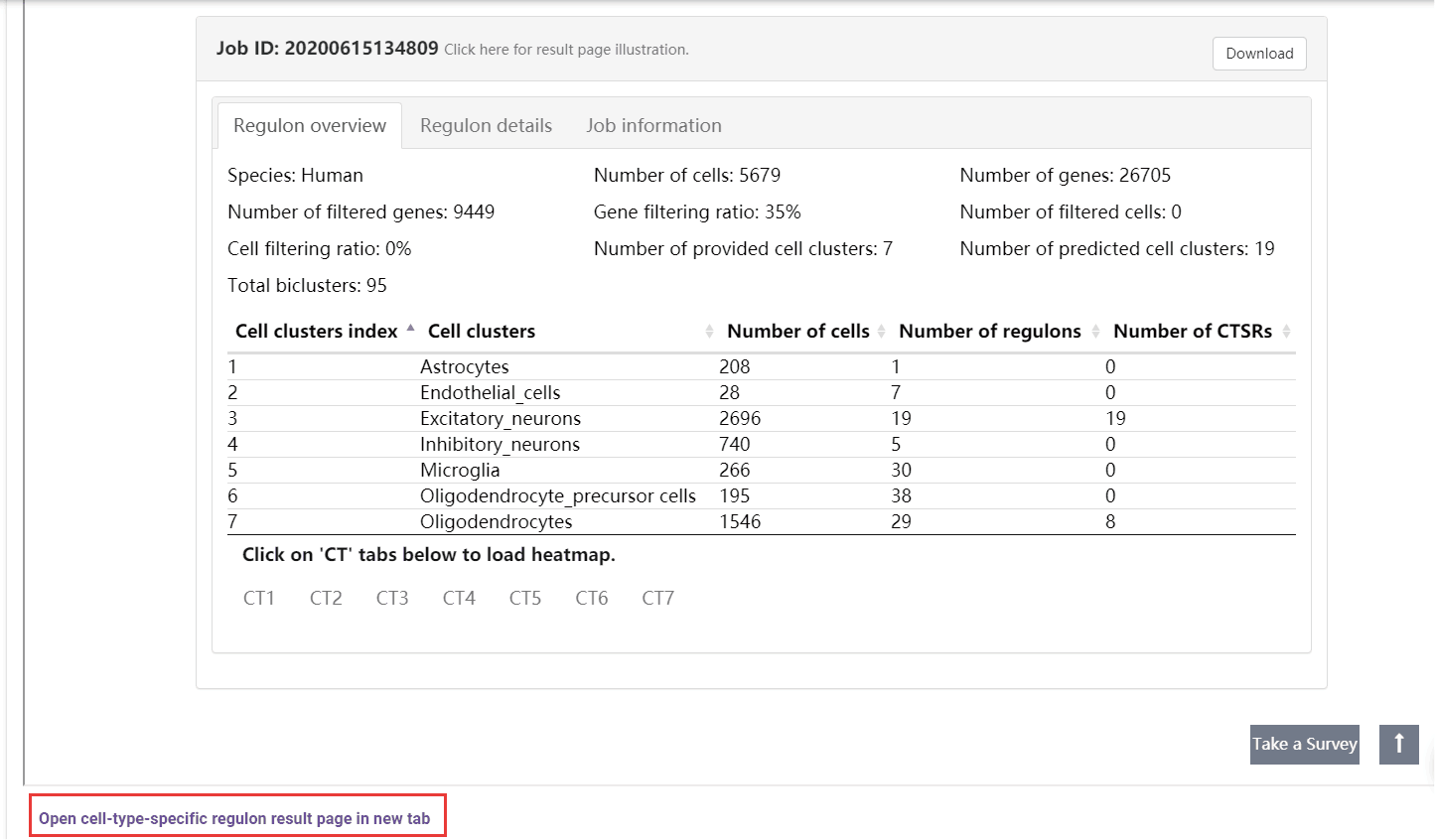
A table summarizes the clusters shows the overall cell number and regulon number in each cluster. Users can jump into the IRIS3 to see the detail results of this job by clicking the ‘Open cell-type-specific regulon result page in new tab’ button. In the table, the index number will be given to represent CTSRs. A cell-gene-regulon heatmap will be displayed for each cell type by clicking on the CT## tab. See the figure details below.
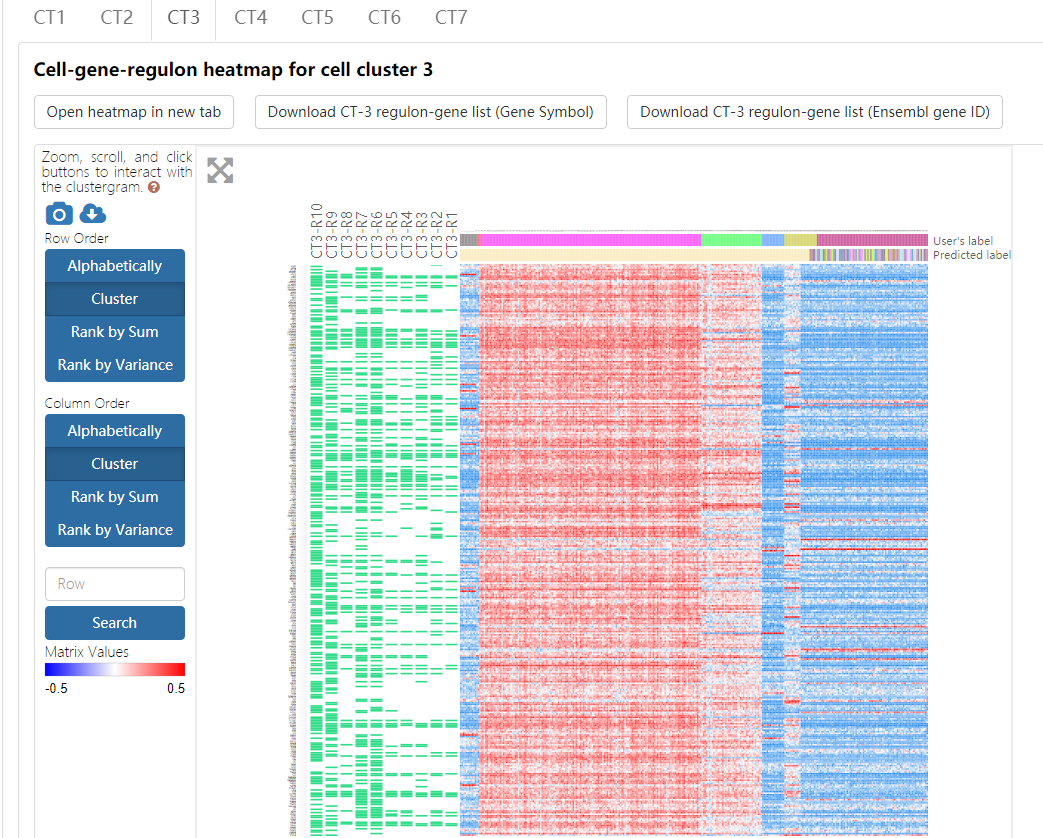
The heatmap, empowered by Clustergrammer, showcases the expression pattern of genes from the top ten regulons in the corresponding cell type. To explore details about Clustergrammer, please read the Clustergrammer documentation.
- Both gene compositions of regulons and their expression values across different cell types can be intuitively displayed in such a heatmap. Regulons are ranked in increasing order of the empirical p-values of regulon specificity scores (RSS) as described above, and a regulon is named as CTn-Rm with n representing the index of cell type and m represents the regulon rank. Due to the space limitation, only the top ten regulons and their corresponding genes are showcased in the heatmap, and the component genes of each regulon are indicated as green rectangles. The heatmap records the log-transformed expression level of each top-ten-regulon-covered gene across all cells.
- Cell names, original cell type labels, and cell types labels predicted by Seurat are shown on the heatmap. This heatmap can also be sorted by gene and cell by double-clicking on the appropriate area on the image. Conveniently, a series of gene enrichment tests can be directly performed on the heatmap using the integrated Enrichr function in the Clustergrammer framework.
- This heatmap can be downloaded by clicking on the top-left buttons above the heatmap and choosing the ‘Take a snapshot’ option.
- Regulon details
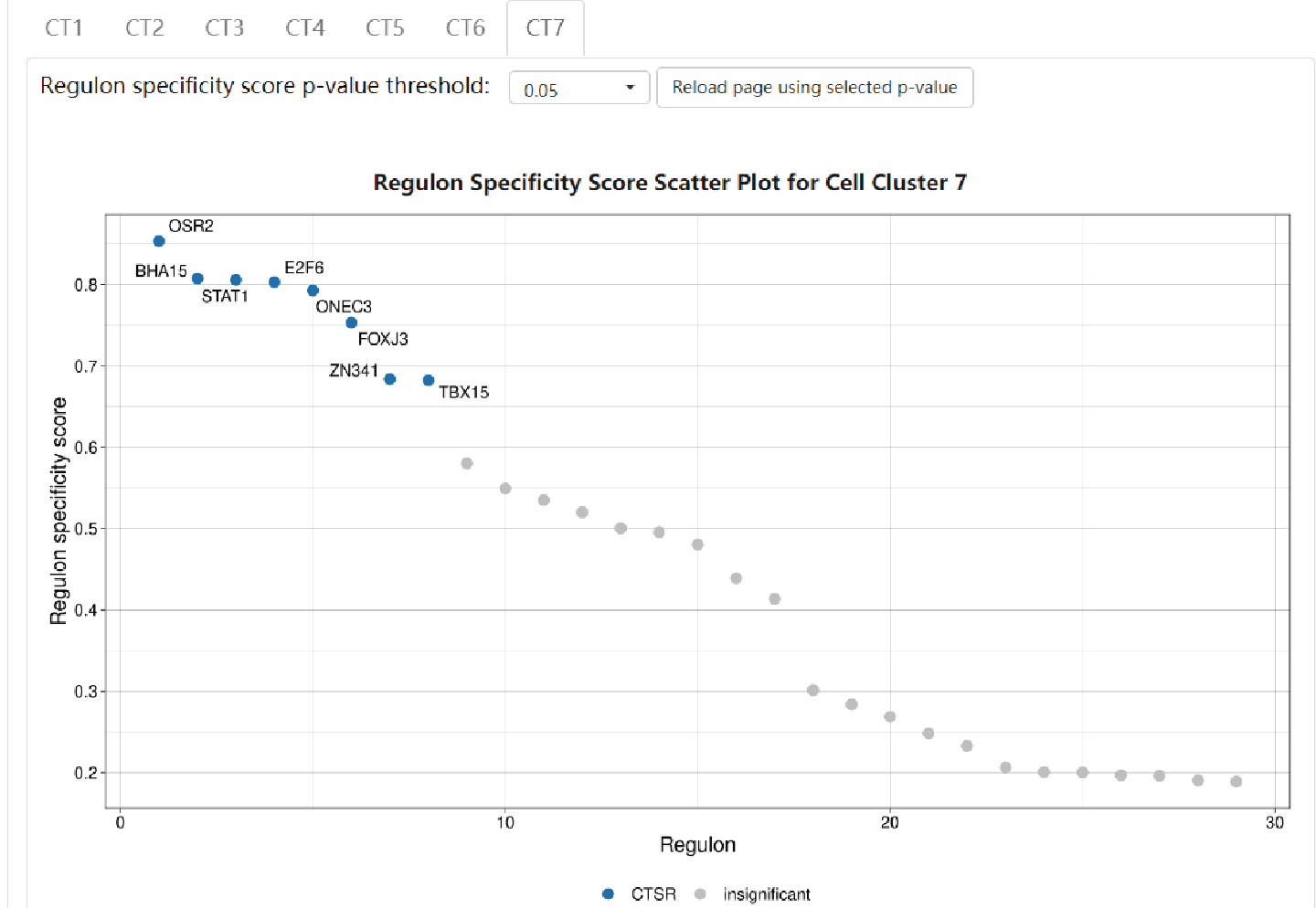
Regulon results are separately showcased in each cell type. Click on the “CT#” button to switch to see results in other cell types. A scatter plot shows the distribution of RSS of each regulon. CTSRs are marked as blue dots with their representative TF names besides, and insignificant regulons are marked as grey dots. Detailed information of each regulon can be found in IRIS3.
What is IRIS3?
IRIS3 (Integrated Cell-type-specific Regulon Inference Server from Single-cell RNA-Seq) is an easy-to-use server empowered by over 20 functionalities to support comprehensive interpretations and graphical visualizations of identified CTSRs. CTSR data can be used to reliably characterize and distinguish the corresponding cell type from others and can be combined with other computational or experimental analyses for biomedical studies. CTSRs can, therefore, aid in the discovery of major regulatory mechanisms and allow reliable constructions of global transcriptional regulation networks encoded in a specific cell type. The broader impact of IRIS3 includes, but is not limited to, investigation of complex diseases hierarchies and heterogeneity, causal gene regulatory network construction, and drug development. IRIS3 is freely accessible from https://bmbl.bmi.osumc.edu/iris3/ with no login requirement. More information about this paper can be found here.
What are the input files of the ssREAD?
The required input file is a gene expression matrix with three acceptable formats, compressed files are encouraged to decrease the uploading time.
- A single txt, tsv or csv formatted gene expression matrix. The compressed format (gzip) is accepted
- An hdf5 feature barcode matrix.
- The three gzipped files recording information of barcodes, features, and gene expressions in the 10X genomics output folder.
Identifiers can be represented as Gene Symbols (e.g., HSPA9), Ensembl Gene IDs (e.g., ENSG00000113013), or Transcript IDs (e.g., ENSMUST00000074805). Human and mouse genes are annotated with respect to the reference genome using the org.HS.eg.db R package and org.Mn.eg.db package, respectively.
How to interpret the file names?
The file name is composed of six parts, they are species, condition, region, gender, age, and code numbers based on the string order of the file_name. Here are two examples showing how to read the file_name column:
| file_name | species | condition | region | gender | age | code_numbers |
|---|---|---|---|---|---|---|
| M-H-Cerebellum-Male-7m | Mouse | Control | Cerebellum | Male | 7 months | NULL |
| H-AD-Prefrontal cortex-Female_001 | Human | Disease | Prefrontal cortex | Female | 75 – 90+ years | 001 |
Note: For control datasets across different brain regions with different genders in different species, each of which corresponded to only one file, so it doesn’t have any code_numbers here.
How to interpret the data_id?
The data_id name is composed of three parts, they are Alzheimer’s disease (the two letters), the region of different species (the first three numbers), and code numbers (the last two numbers).
Here is the table showing which brain regions are the first three letters corresponded to:
| Three letters | Brain Region |
|---|---|
| 001 | Human prefrontal cortex |
| 002 | Human entorhinal cortex |
| 003 | Mouse cortex |
| 004 | Mouse cerebral cortex |
| 005 | Mouse cerebellum |
| 006 | Mouse prefrontal cortex |
| 007 | Mouse hippocampus |
| 008 | Human superior frontal gyrus |
| 009 | Mouse cortex and hippocampus |
| 010 | Mouse subventricular zone and hippocampus |
| 011 | Human prefrontal cortex (BA9) |
| 012 | Human superior parietal lobe |
Note: We separated the mouse brain region of cortex from the cerebral cortex because the corresponding datasets used different mouse models. We separated the human prefrontal cortex brain region because the corresponding braak stages are different.
What is fst format?
Multithreaded serialization of compressed data frames using the ‘fst’ format. The ‘fst’ format allows for random access of stored data and compression with the LZ4 and ZSTD compressors created by Yann Collet.
You can see more information about Package ‘fst’ here.
Where can I find the source code?
The source code of ssREAD can be accessed on GitHub.
What are the future plans of ssREAD?
- Collect more AD scRNA-Seq, snRNA-Seq data, spatial transcriptomics from more brain regions, and build up healthy atlas in diverse brain regions of human, mouse and extend to other species.
- Collect AD single-cell omics data, such as scATAC-seq data, and achieve more comprehensive analysis results based on single-cell multiple omics data.
What computational tools are used in ssREAD?
| Tools | Publication |
|---|---|
| IRIS3 | Anjun Ma, Cankun Wang, Yuzhou Chang, Faith H Brennan, Adam McDermaid, Bingqiang Liu, Chi Zhang, Phillip G Popovich, Qin Ma, IRIS3: integrated cell-type-specific regulon inference server from single-cell RNA-Seq, Nucleic Acids Research, gkaa394, https://doi.org/10.1093/nar/gkaa394 |
| Seurat | Stuart T*, Butler A*, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R. Comprehensive Integration of Single-Cell Data Cell. 2019. DOI:https://doi.org/10.1016/j.cell.2019.05.031 |
| Harmony | Korsunsky, I., Millard, N., Fan, J. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296 (2019). https://doi.org/10.1038/s41592-019-0619-0 |
| Polychrome | Coombes, K., Brock, G., Abrams, Z., & Abruzzo, L. (2019). Polychrome: Creating and Assessing Qualitative Palettes with Many Colors. Journal of Statistical Software, 90(Code Snippet 1), 1 - 23. doi:http://dx.doi.org/10.18637/jss.v090.c01 |
| Destiny | Angerer P, Haghverdi L, Büttner M, Theis F, Marr C, Büttner F (2015). “destiny: diffusion maps for large-scale single-cell data in R.” Bioinformatics, 32(8), 1241 1242 1243. doi: 10.1093/bioinformatics/btv715, More information at http://www.helmholtz-muenchen.de/icb/destiny, http://bioinformatics.oxfordjournals.org/content/32/8/1241. |
| SCINA | Zhang, Z., Luo, D., Zhong, X., Choi, J. H., Ma, Y., Wang, S., Mahrt, E., Guo, W., Stawiski, E. W., Modrusan, Z., Seshagiri, S., Kapur, P., Hon, G. C., Brugarolas, J., & Wang, T. (2019). Scina: Semi-supervised analysis of single cells in silico. Genes, 10(7), [531]. https://doi.org/10.3390/genes10070531 |
Why does ssREAD use PCA instead of CCA or LSI for the dimensional reduction in the identification of disease dataset cell types?
In the identification of disease cell types, we used Seurat’s FindTransferAnchors and TransferData function to transfer cell types from the control atlas to disease datasets, which project PCA structure from the query (disease dataset) onto the reference (control atlas). PCA is recommended when reference and query datasets are from scRNA-seq. CCA is considered better captures the shared feature correlation structure across single-cell modalities. LSI is considered better captures the ‘internal’ structure of ATAC-seq data in the case of scRNA-seq to scATAC-seq transfer.
What is ssREAD’s scRNAseq pipeline?
Our database organically integrates multiple state-of-the-art tools, including IRIS3, SCINA, Seurat, and IRIS-EDA. These tools are either reputed or have been justified with the best performance in the analytical area.
1. Construction of Human and Mouse control atlas
Human and Mouse control atlases come from the 15 scRNA-Seq & snRNA-Seq studies. Genes detected in less than 3 cells and cells detected in less than 200 genes were filtered out. Principal component analysis (PCA) was performed to obtain a small number of principal components, 25 PCA components were used as input of Uniform Manifold Approximation and Projection (UMAP). Initial clustering was performed using Seurat’s v3.1.5 SNN graph clustering using the FindClusters function with a resolution of 0.8.
SCINA is an R package that leverages prior marker genes information and simultaneously performs cell type clustering and assignment for known cell types, SCINA shows top performances among prior-knowledge classifiers when high-quality marker genes were provided. Each cell was assigned a cell type based on a manually created marker gene list file using SCINA v1.2.0, the cells with unknown labels marked by SCINA were first compared with predicted clusters from Seurat, then the unknown labels were assigned to the most dominate cell types within the predicted clusters.
Marker genes
We found some of the marker genes used in v1 were not specific enough during the development of ssREAD. To improve the cell type annotate accuracy, we filtered out several markers and used an updated workflow by performing two iterations of cell type annotation. The differences are that only Neurons would be annotated during the first iteration. Next, the Neurons were a subset from the whole dataset and used iteration 2 marker genes to annotate the Excitatory neurons and Inhibitory neurons. Finally, the Neurons’ cell labels would be replaced by Excitatory neurons or Inhibitory neurons. The new workflow can improve the annotation by taking considering the differences between cell types and subtypes.
Iteration 1 marker genes:
| Astrocytes | Endothelial cells | Neurons | Microglia | Oligodendrocytes | Oligodendrocyte precursor cells | Pericytes |
|---|---|---|---|---|---|---|
| GFAP | CLDN5 | GLS | P2RY12 | MBP | VCAN | AMBP |
| AQP4 | VWF | RBFOX3 | CSF1R | MOBP | SOX8 | HIGD1B |
| GJA1 | CAMK2A | CX3CR1 | PLP1 | PTH1R | ||
| SLC1A2 | C3 | MYRF | ||||
| FGFR3 | MAG | |||||
| NKAIN4 | ||||||
| AGT | ||||||
| PLXNB1 | ||||||
| SLC1A3 |
Iteration 2 marker genes:
| Excitatory neurons | Inhibitory neurons |
|---|---|
| SLC17A6 | GAD1 |
| SLC17A7 | GAD2 |
| SATB2 |
2. Identification of Human and Mouse disease cell types based on the control atlas
Not all cells collected from patient samples are malignant, there are heterogeneous cells within individual patients, that is, normal healthy cells are included. In Granja et al.’s research, they defined these healthy cells as control-like cells. These control-like cells maintain distinct regulatory mechanisms and gene expression patterns compared to disease cells and will disturb the accurate identification of cancer cell clusters. Thus, the removal of control-like cells from disease data is critical to identify real disease-associated cells. Granja et al. used this strategy to remove control-like cells, and we used this strategy in ssREAD to identify AD-associated cells. For each of the AD datasets in ssREAD, the ratio of the control-like cells out of all the cells in this dataset is about 10%. We tested at Mathys et al.’s dataset, and found out the ARI scores between with control-like cells and without control-like cells has no significant difference. However, the ARI score of without control-like cells datasets is higher than with control-like cells datasets.
To determine whether cells from disease datasets are control-like, Harmony R package v1.0 was first used to integrate the disease dataset with its corresponding control atlas. Harmony shows similar ARI scores but having a significantly shorter run-time compared to other data integration tools. After the integration, cells were clustered using Seurat FindClusters function with a resolution of 4. A hypergeometric test was performed for each cluster using the number of cells from disease cells and the number of cells from the control atlas. Clusters were considered to be control-like if the hypergeometric test result was significant (p-value < 0.0001, Benjamini-Hochberg adjusted), and the cells from the disease dataset in control-like clusters were removed from the downstream analyses.
For the remaining disease cells, Seurat’s FindTransferAnchors function was used to find transfer anchors using PCA to project the control-atlas onto the disease dataset. Cell types were transferred using the TransferData function with PCA embeddings as the weighting anchors. The subclusters for each cell types were designated using Seurat FindClusters function for all cells in each identified cell type with a resolution of 0.8.
3. Identification of differentially expressed genes
Seurat’s FindAllMarkers and FindMarkers functions that utilize the MAST package were used to run DE testing on normalized gene expression data. Cell-type-specific genes were identified by performing DE testing between the cell type of interest and the average of the remaining cell types. Subcluster-specific genes were identified by performing DE testing between the subcluster of interest and the average of the remaining subclusters from the same cell type. For each cell type, several DE comparisons were performed within two different datasets, categorized from AD versus control, and AD versus AD in the same species under the same gender, brain region, and age. To regress out technical biases from different datasets, the dataset latent variables were added in all cross-dataset DE testing. All of the above-mentioned DE results can be sent to Enrichr web server real-time against different functional annotation databases to identify the enriched KEGG pathways, Gene Ontology(GO), etc.
4. Identification of CTSRs
Cell-type-specific regulons (CTSRs) are defined as a group of genes, which receive similar regulatory signals in a specific cell type, hence tending to have similar expression patterns and share conserved motifs in this cell type. A successful elucidation of CTSRs will substantially improve the identification of transcriptionally co-regulated gene modules, realistically allowing reliable prediction of global transcription regulation networks encoded in a specific cell typeThe CTSRs analysis in ssREAD is performed using default IRIS3 settings.
What is ssREAD’s spatial transcriptomics pipeline?
1. Data preprocessing
For each samples within the study, we imported them into the Squidpy Python package. We follow the standard workflow on Squidpy and Scanpy.
2. Identification of Spatially variable genes (SVGs)
SpaGCN is a graph convolutional network model. We used SpaGCN to integrate gene expression and histology to identify spatial domains and spatially variable genes.
3. Identification of differentially expressed genes
Seurat’s FindAllMarkers and FindMarkers functions that utilize the MAST package were used to run DE testing on normalized gene expression data. Cell-type-specific genes were identified by performing DE testing between the cell type of interest and the average of the remaining cell types. Subcluster-specific genes were identified by performing DE testing between the subcluster of interest and the average of the remaining subclusters from the same cell type. For each cell type, several DE comparisons were performed within two different datasets, categorized from AD versus control, and AD versus AD in the same species under the same gender, brain region, and age. To regress out technical biases from different datasets, the dataset latent variables were added in all cross-dataset DE testing. All of the above-mentioned DE results can be sent to Enrichr web server real-time against different functional annotation databases to identify the enriched KEGG pathways, Gene Ontology(GO), etc.
4. Deconvolution analysis with scRNA-seq & snRNA-seq datasets
CARD is a reference-based deconvolution method that estimates cell type composition in spatial transcriptomics based on cell type specific expression information obtained from a reference scRNA-seq data. In ssREAD, we used CARD to track compositional alterations of cell types (from scRNA-seq or snRNA-seq dataset) in gene expression data (spatial transcriptomics dataset).
5. Exporting sample ojbects for Python & R environment
For each samples within the study, we provite raw and processed data object. The objects are both exported the object to h5ad format for Python users, and h5Seurat object for R users. The seurat-disk package was used to convert the h5ad object to h5Seurat format.